Reinraumprojekte: Herausforderung für Bauherren und Projektsteuerer | Beitrag aus cleanroom & processes 3 | Nr. 4 | 196-203 (2024)
20.12.2024Viele Hightech-Produkte, wie Halbleiter oder pharmazeutische Produkte, erfordern extreme Reinheit, um sicherzustellen, dass sie korrekt funktionieren oder wirksam sind. In der Medizin kann eine...

SCHOTT Pharma erreicht starkes Wachstum und Rekordmarge nach gutem Jahresendspurt
19.12.2024Alle Ziele für 2024 erreicht, einschließlich angehobener Umsatzprognose: 12 % Umsatzwachstum und...
Wechsel an der Spitze von Endress+Hauser InfoServe
18.12.2024Oliver Blum übernimmt die Geschäftsführung des gruppenweiten IT-Dienstleisters...
Lonza has provided an overview of its new “One Lonza” strategy and new organizational structure...
17.12.2024Lonza has provided an overview of its new “One Lonza” strategy and new organizational structure...
Branchenpartner


meistgelesen

Beitrag aus der Ausgabe 4/2024 der Zeitschrift cleanroom & processes
An overview of sterility testing
The impact of Annex 1 and a look into the future of sterility testing
Sterility testing is a process which must be performed as part of the manufacture of sterile products, to provide confidence that they are free of any viable microorganisms which could harm patients. As it is not possible to test every single vial or ampoule of product that is being manufactured, a number of samples representative of the whole batch are taken at separate times during the filling operation and tested for microbial contamination. ...
Beitrag aus der Ausgabe 4/2024 der Zeitschrift cleanroom & processes
Herstellung steriler Arzneiformen
Zwischen Technologiefortschritt und wachsenden regulatorischen Anforderungen
Die Herstellung von Arzneimitteln hat unter definierten und kontrollierten Umgebungsbedingungen i. d. R. in Reinräumen zu erfolgen. Reinräume bilden ein zentrales Element der Herstellung unter Good-Manufacturing-Practice(GMP)-Bedingungen. Je nach herzustellender Darreichungsform und Applikationsroute des resultierenden Arzneimittels ist die Reinheitsklasse der Reinräume zu wählen. Für orale und topische Darreichungsformen gelten ...

Beitrag aus der Ausgabe 4/2024 der Zeitschrift cleanroom & processes
Heading for the Green Cleanroom
Simulation-Based Approach for Energy Optimization
A comprehensive view on production systems and their infrastructure is essential for pharmaceutical companies to achieve their decarbonization and sustainability objectives. Cleanroom technology is a particularly encouraging place to start when it comes to energy optimization. This article introduces a new methodology that has been developed for process-based, energy-optimized cleanroom planning, in which simulations play a central role.Process ...
Top Themen

Beitrag aus der Ausgabe 4/2024 der Zeitschrift cleanroom & processes
Learning Solution für Mitarbeiterqualifizierung im Kontext Reinraum und CCS
Blended Learning als essenzieller Baustein
Wenn man den Begriff Human Resources (HR) hört, ist gerade die Bezeichnung Ressource oft negativ konnotiert oder wird abgelehnt. Aber die Mitarbeiterinnen und Mitarbeiter eines Unternehmens sind schon lange nicht mehr nur Arbeitskräfte. Wie auch Ressourcen zeichnen sich Beschäftigte dadurch aus, dass sie wertvoll, rar, nicht imitierbar und nicht einfach ersetzbar sind. Der Mensch im Unternehmen wird als höchst wertvolle Ressource gesehen, weil ...


Vorschau (Änderungen vorbehalten)

Beitrag aus der nächsten Ausgabe 01/2025 der Zeitschrift cleanroom & processes
(erscheint am 07.03.2025)
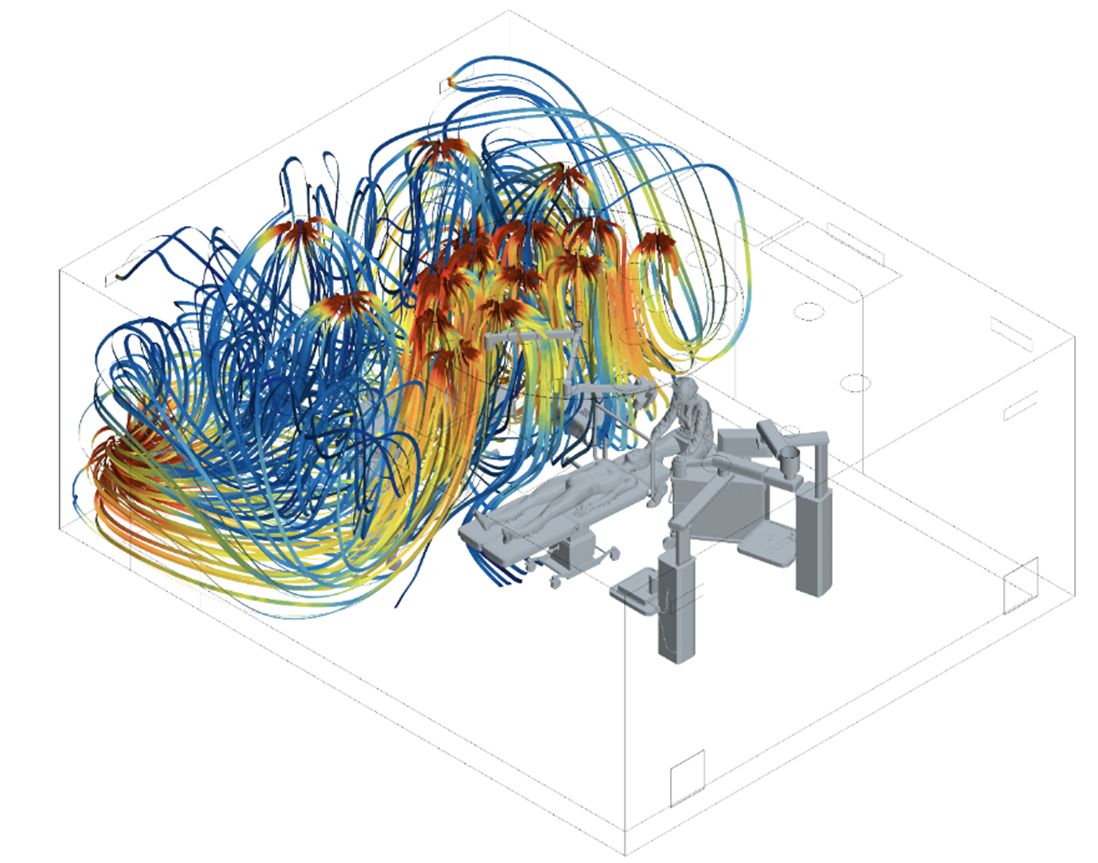
CFD-Simulation zum Vergleich von Deckenumluftgeräten mit traditionellen Drallauslässen und bodennaher Absaugung
Die Studie vergleicht mithilfe von CFD-Simulationen Deckenumluftgeräte mit herkömmlichen Lüftungsanlagen in einem Reinraum, wobei der Fokus auf Temperaturverteilung, Partikelgeschwindigkeit und Erholzeiten liegt. Deckenumluftgeräte sind modular aufgebaut und arbeiten im Umluftbetrieb, was eine effiziente Luftzirkulation ermöglicht. Die Simulation zeigt, dass diese Geräte Vorteile bei der Verteilung von Partikelgeschwindigkeit und Temperatur haben, da sie aufsteigende Warmluft direkt an der Decke abführen und so Wärmestau vermeiden. Beide Systeme sorgen für ausreichende Luftbewegung im Raum, auch in Totzonen. Die Strömungsgeschwindigkeiten der Deckenumluftgeräte übertreffen die Minimalanforderungen, was eine schnelle Partikelreduktion ermöglicht. Die Erholzeit zur Reduzierung der Partikelkonzentration beträgt in beiden Systemen unter 20 min. Die Ergebnisse bestätigen die Effektivität der Deckenumluftgeräte und die Bedeutung von CFD-Simulationen zur Optimierung von Lüftungssystemen. Deckenumluftgeräte sind mindestens gleichwertig mit herkömmlichen Systemen und bieten eine solide Basis für ihre Weiterentwicklung in Reinraumanwendungen.