



Visionäre bionisch inspirierte Leichtbauten für modulare Tragstrukturen, Beitrag aus cleanroom & processes 3, Nr. 1, 24-30 (2024)
02.05.2024Modulare Reinraumkonzepte gewinnen in der pharmazeutischen Industrie zunehmend an Beliebtheit, da sie zu höherer Flexibilität, Qualität, Sicherheit und Nachhaltigkeit der Anlagen beitragen. Dabei ...

Das umfangreiche Messgeräteprogramm für maximale Sicherheit im hygienischen Prozess
07.05.2024Spezifische Eigenschaften von Produkten in der Chemie-, Pharma- und Nahrungsmittelindustrie setzen die Einhaltung besonderer Hygieneanforderungen bei ihrer Verarbeitung in hochkomplexen, teilweise ...
Podcast Reingehört Spezial #1: Steffen Röhm, Weiss Technik, über Luftentfeuchtung im reinen Umfeld
07.05.2024Steffen Röhm von Weiss Technik enthüllt die Herausforderungen und Lösungen der Luftentfeuchtung für Industrieprozesse, von der Batterieproduktion bis zur Brauseherstellung. Beleuchtet werden die ...

Erbe Elektromedizin eröffnet neues Kompetenzzentrum für medizintechnische Instrumente in Rangendingen und erweitert seine Kapazitäten für Kunden in aller Welt
06.05.2024Nach drei Jahren Bauzeit und einer Gesamtinvestition von 90 Millionen Euro hat am 3. Mai das neues Produktions- und Entwicklungswerk in Rangendingen bei Tübingen eröffnet. Dort werden...
Branchenpartner

Top Downloads
Beitrag aus der Ausgabe 1/2024 der Zeitschrift cleanroom & processes
Annex 1 – Handlungsempfehlungen für eine GMP-konforme und effiziente Reinraummesstechnik
Brewi et al. • Annex 1 – Reinraummesstechnik
Die im aktuellen Annex 1 des EU-Leitfadens zur Good Manufacturing Practice (GMP) – gültig seit 25.08.2023 – festgelegten Umgebungsbedingungen, Mindestanforderungen für die Qualifizierung und Prüfintervalle für die Requalifizierung sind einzuhalten:Umgebungsbedingungen in Reinräumen, Schleusen und Durchreichen: z. B. lebensfähige und nicht vermehrungsfähige Partikel, Luftströmung, ÜberdruckMindestanforderungen für die ...
Beitrag aus der Ausgabe 1/2024 der Zeitschrift cleanroom & processes
Schutz von kritischen Komponenten vor Oberflächenkontaminationen
Teil 1
In allen Bereichen kommt es, je nach Umgebungsbedingungen, zur zeitlichen Ablagerung von luftgetragenen Kontaminationen. Dies gilt auch für kontrollierte Umgebungsbedingungen – wie in Reinräumen. Die Luft in Reinräumen wird zwar gefiltert und überwacht; trotzdem lagern sich auch hier über die Zeit Kontaminationen auf kritischen Komponenten, Produkten und Oberflächen ab. Daher ist es gängige Praxis, diese je nach Prozessanforderungen mit ...
Beitrag aus der Ausgabe 1/2024 der Zeitschrift cleanroom & processes
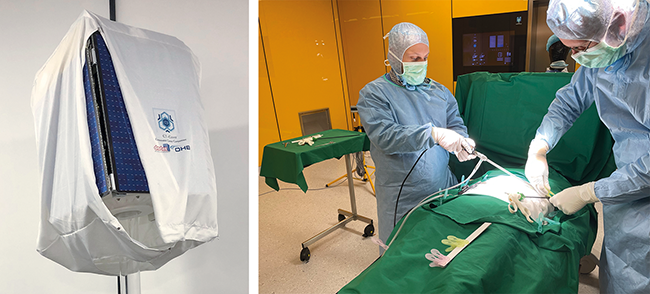
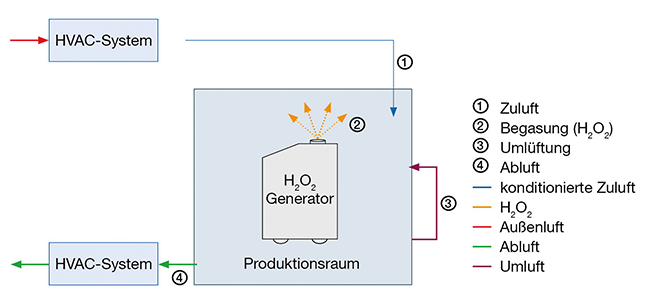
Effektive Reinraum-Dekontamination mit Wasserstoffperoxid
Durchführungsweise, Systemqualifizierung, Zyklusentwicklung und -validierung
Die Herstellung steriler Produkte erfordert spezielle Produktions- und Umgebungsanforderungen, um sicherzustellen, dass der Herstellungsprozess den geltenden gesetzlichen Vorschriften und Richtlinien sowie dem aktuellen Stand der Wissenschaft und Technik entspricht [1–3]. Die Gewährleistung einer optimalen Produktionsumgebung ist von entscheidender Bedeutung, um eine qualitativ hochwertige und sichere Herstellung von Produkten zu ...
Top Themen
Beitrag aus der Ausgabe 1/2024 der Zeitschrift cleanroom & processes
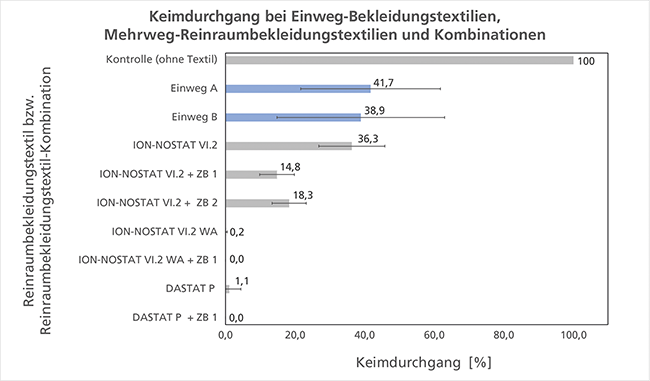
Bewährungsprobe für Reinraumbekleidung
Die ReBa2-Prüfmethode zur Bestimmung des Keimdurchgangs – Teil 2*Teil 1 dieses Beitrags s. cleanroom & processes 2023;2(4): 174–178.
Die quadratischen Messproben werden zugeschnitten, die Außen- bzw. Innenseite markiert und sterilisiert. Das Sterilisationsverfahren wird vorzugsweise entsprechend der geplanten Anwendung ausgewählt, z. B. Dampfsterilisation.Bei Materialien, die bereits in sterilem Zustand geliefert werden, ist eine aseptische Messprobenentnahme möglich.Die Komponenten Baumwollgewebe, Keimträger- sowie Abdeckfolie werden dampfsterilisiert.Das Prüfgerät ...


Vorschau (Änderungen vorbehalten)
Beitrag aus der nächsten Ausgabe 02/2024 der Zeitschrift cleanroom & processes
(erscheint am 31.05.2024)
Realisierung von Gefriertrocknungsanlagen / Aktuelle regulatorische Anforderungen an Reinraumtechnologien zur aseptischen Prozessführung – Teil 1
Der Prozess der Gefriertrocknung, auch Lyophilisation genannt, nimmt in pharmazeutischen Herstellungsprozessen eine tragende Rolle bei der Stabilität von sensitiven Arzneimitteln ein. Für die Umsetzung der regulatorischen Anforderungen ist im Zusammenhang mit der Gefriertrocknung von sterilen Arzneimitteln insbesondere der Annex 1 zum Leitfaden der EU zur Good Manufacturing Practice (GMP) in der Fassung vom 25. Aug. 2022 zu berücksichtigen. Dieser Beitrag beschreibt die Anforderungen des Annex 1 an die gebotenen Reinraumtechnologien im Kontext mit Gefriertrocknungsanlagen. Darüber hinaus wird aufgezeigt, worauf geachtet werden muss, um Gefriertrocknungsprozesse entsprechend den GMP-Vorgaben in aseptischen Umgebungen durchführen zu können.






