
Privatsphäre-Einstellungen
Wir verwenden Cookies auf unserer Website. Einige von ihnen sind unerlässlich, während andere uns helfen, diese Website und Ihre Erfahrungen zu verbessern.
Hier finden Sie eine Übersicht aller verwendeten Cookies. Sie können ganzen Kategorien Ihre Zustimmung geben oder weitere Informationen anzeigen und bestimmte Cookies auswählen.
| Notwendig Cookies | Weitere Informationen | |
| Wesentliche Cookies ermöglichen grundlegende Funktionen und sind für die ordnungsgemäße Funktion der Website erforderlich. | ||
| Name | Anbieter | Zweck | Ablauf |
|---|---|---|---|
| cb_enabled | ecv.de | Mit diesem Cookie wird festgestellt, ob der Besucher die Cookie-Zustimmungsbox akzeptiert hat | 1 Jahr |
| PHPSESSID | ecv.de | Erhält den Benutzersitzungsstatus über Seitenanforderungen hinweg | Session |
| ECVRL | ecv.de | Mit diesem Cookie wird für die Website eine Möglichkeit geschaffen den Besucher auch zu einem späteren Besuch wieder zu erkennen und so wichtige Funktionen wie die Suche/Merkzettel/Shop funktionell durchführen zu können. | 1 Jahr |
| Statistik Cookies | Weitere Informationen | |
| Statistik-Cookies sammeln anonym Informationen. Diese Informationen helfen uns zu verstehen, wie unsere Besucher unsere Website nutzen. | ||
| Name | Anbieter | Zweck | Ablauf |
|---|---|---|---|
| utm.gif | www.google.com | Google Analytics-Tracking-Code, der Details zum Browser und Computer des Besuchers protokolliert. | Session |
| _utma | ecv.de | Sammelt Daten darüber, wie oft ein Benutzer die Website besucht hat, sowie Daten für die erste und die meisten letzter Besuch. Wird von Google Analytics verwendet. | 2 Jahre |
| __utmb | ecv.de | Registriert einen Zeitstempel mit dem genauen Zeitpunkt, zu dem der Benutzer auf die Website zugegriffen hat. Wird von Google Analytics verwendet, um die Dauer eines Website-Besuchs zu berechnen. | 1 Tag |
| __utmc | ecv.de | Registriert einen Zeitstempel mit dem genauen Zeitpunkt, zu dem der Benutzer auf die Website zugegriffen hat. Wird von Google Analytics verwendet, um die Dauer eines Website-Besuchs zu berechnen. | 1 Tag |
| __utmt | ecv.de | Wird verwendet, um die Geschwindigkeit von Anforderungen an den Server zu drosseln | 1 Tag |
| __utmz | ecv.de | Sammelt Daten darüber, woher der Benutzer kam, welche Suchmaschine verwendet wurde, auf welchen Link geklickt wurde und welcher Suchbegriff verwendet wurde. Wird von Google Analytics verwendet. | 6 Monate |
| Marketing Cookies | Weitere Informationen | |
| Marketing-Cookies werden von Werbekunden oder Publishern von Drittanbietern verwendet, um personalisierte Anzeigen zu schalten. Sie tun dies, indem sie Besucher über Websites hinweg verfolgen | ||
| Name | Anbieter | Zweck | Ablauf |
|---|---|---|---|
| _session_id | delivery.selfcampaign.com | Speichert die Navigation der Besucher durch Registrieren von Zielseiten - Auf diese Weise kann die Website relevante Produkte präsentieren und / oder ihre Werbeeffizienz auf anderen Websites messen. | Session |
| _session_id | lib.selfcampaign.com | Speichert die Navigation der Besucher durch Registrieren von Zielseiten - Auf diese Weise kann die Website relevante Produkte präsentieren und / oder ihre Werbeeffizienz auf anderen Websites messen. | Session |
| axd | theadex.com | Legt eine eindeutige ID für den Besucher fest, mit der Werbetreibende von Drittanbietern den Besucher mit relevanter Werbung ansprechen können. Dieser Pairing-Service wird von Werbe-Hubs von Drittanbietern bereitgestellt, mit dem Werbung in Echtzeit angeboten werden kann. | 1 Jahr |
| d/cm.gif | theadex.com | Präsentiert dem Benutzer relevante Inhalte und Werbung. Der Service wird von Dritten über Werbe-Hubs eingestellt, die das Anzeigen von Werbung in Echtzeit ermöglichen. | Session |
| IDE | doubleclick.net | Wird von Google Doubleclik verwendet,um die Aktionen des Website-Nutzers nach dem Anzeigen oder Klicken auf eine Anzeige des Werbetreibenden zu registrieren und zu melden, um die Wirksamkeit einer Anzeige zu messen und dem Nutzer gezielte Anzeigen zu präsentieren. | 1 Jahr |
| TDCPM | adsrvr.org | Registriert eine eindeutige ID, die das Gerät eines zurückkehrenden Benutzers identifiziert. Die ID wird für gezielte Anzeigen verwendet. | 1 Jahr |
| uid | adform.net | Registriert eine eindeutige Benutzer-ID, die den Browser des Benutzers beim Besuch von Websites erkennt, die dasselbe Anzeigennetzwerk verwenden. Ziel ist es, die Anzeige von Anzeigen basierend auf den Bewegungen des Nutzers und den Geboten verschiedener Anzeigenanbieter für die Anzeige von Nutzeranzeigen zu optimieren. | 2 Monate |
| _li_id_3004 | leadinfo | Speichert das Nutzerverhalten der Benutzer der Website | 2 Monate |


|
Herzlich Willkommen im Auditorium! Im Auditorium unserer Fachzeitschrift cleanroom & processes publizieren wir zusätzliche Beiträge von Unternehmen und Autoren aus der Pharmaindustrie. Diese Beiträge durchlaufen kein Peer Review. Für die Qualität der Inhalte steht der jeweilige Autor. Bei Fragen nutzen Sie bitte seine Kontaktadresse am Beitragsende. |
| Newsletter | ||
| Newsletter Juli 2023 Innovative Dichtheitsprüfung pharmazeutischer Verpackungen: Der AMI von Pfeiffer Vacuum |
| |
Pfeiffer Vacuum GmbH, 5. Juli 2023Die Qualität und Wirksamkeit von Medikamenten, hängt entscheidend mit ihrer Verpackung zusammen. Diese muss unversehrt und von höchster Güte sein, andernfalls kann es zu schwerwiegenden Folgen kommen. Ein Fall aus den frühen 1970er Jahren belegt das: Damals kam es in den USA aufgrund von verunreinigten Fluiden zu schätzungsweise 2.000 – 8.000 Infektionen der Blutgefäße, die bei rund 10% der Erkrankten zum Tod führten. Dieses massive Versagen des Verpackungsverbunds hat ein gesteigertes Bewusstsein für die Bedeutung der Qualität pharmazeutischer Verpackungen erzeugt. In den frühen 1980er Jahren wurden Aspekte der Verpackungsdichtheit (CCI) beschrieben. Physikalische und mechanische Eigenschaften eines Systems aus Schraubdeckelglas und Verschluss wurden dargestellt sowie Ansätze für die Prüfung der Integrität von Dichtungen vorgeschlagen. Diese mikrobiologischen Methoden wurden von vielen Anwendern in der pharmazeutischen Industrie und von pharmazeutischen Aufsichtsbehörden als inoffizieller Standard zur Verifizierung der Unversehrtheit eines sterilen Produkt-Verpackungssystems akzeptiert. In den 1990er Jahren wurde dann eine umfangreiche Liste verschiedener physikalisch-chemischer und mikrobiologischer Testmethoden für die Überprüfung der Verpackungsdichtheit erstellt. Der Bericht enthielt auch die Empfehlung, eine physikalisch-chemische Methode anhand eines direkten Vergleichs mit einer mikrobiologischen Eindringprüfung zu validieren.
Probabilistische versus deterministische TestmethodenMikrobiologische Eindringprüfungen sind probabilistische Testmethoden. Diese Testmethoden basieren auf einer Anzahl aufeinanderfolgender oder gleichzeitiger Ereignisse, von denen jedes zufällige Ergebnisse liefert. Diese Ergebnisse sind mit Unsicherheiten behaftet, die eine hohe Anzahl an Experimenten und eine präzise Überwachung der experimentellen Parameter erfordern. Eine mikrobiologische Eindringprüfung kann sehr empfindlich sein und Leckkanäle mit den Abmessungen eines einzelnen Mikroorganismus aufspüren. Klinische Studien haben jedoch nachgewiesen, dass diese Art der Prüfung auch Lecks übersehen kann, welche die Sterilität der Verpackung durchaus gefährden könnten. Demnach ist die Anwendung einer deterministischen Prüfmethode empfehlenswert. Hier basieren die Messungen auf einer vorhersagbaren Kette von Ereignissen. Ein Beispiel für eine deterministische Prüfmethode ist die Helium-Lecksuche.
Helium-Lecksuche bei hermetisch dichten ObjektenDie Helium-Lecksuche an hermetisch dichten Objekten wie pharmazeutischen Verpackungen erfordert eine spezielle Vorbereitung der Verpackung mit Zugabe des Prüfgases Helium. Helium kann mit verschiedenen Verfahren in die Packung eingebracht werden: Befüllen vor VerschließenDiese Methode erfordert die Abdichtung der Verpackung in einer speziellen Atmosphäre, die das Prüfgas Helium enthält. Dies kann beispielsweise in einer eingehausten Station in einer Produktionsstraße oder einem Handschuhkasten für absatzweise Produktion erfolgen. Während des Verschließens muss die Konzentration des Prüfgases Helium präzise überwacht werden, um quantitative Aussagen auf Basis einer bekannten Heliumkonzentration im Inneren des Prüfkörpers zu ermöglichen. Drucklagerungsprüfung (Bombing-Test)In einem ersten Schritt wird hier das zu prüfende Objekt Helium unter hohem Druck in einer Druckkammer, der sogenannten Bombingkammer, ausgesetzt. Das Prüfgas dringt dann durch eventuell vorhandene Leckkanäle in das innere freie Volumen des Prüfobjekts ein. In einem zweiten Schritt wird das Bauteil in einer Vakuumkammer geprüft, die an ein Helium-Lecksuchgerät angeschlossen ist. Die Theorie dieser Methode hat sich etabliert und ist als quantitative integrale Methode klassifiziert.
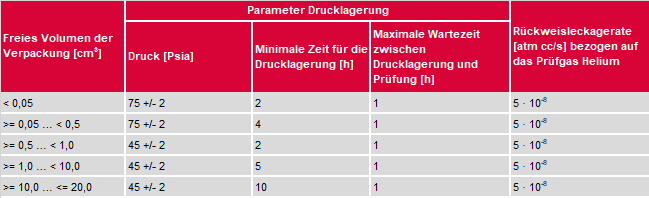
Grenzen der Helium-Dichtheitsprüfung geschlossener BauteileDie Helium-Dichtheitsprüfung hermetisch geschlossener Bauteile unterliegt einigen Einschränkungen. Während der Lagerung der Prüfobjekte in der Druckkammer muss das Prüfgas durch enge Leckagen diffundieren. Die Heliumkonzentration im Inneren des Prüfobjekts folgt einer exponentiellen Zunahme. Der Vorgang ist dabei umso langsamer, je kleiner die Dimensionen des Lecks sind. Oftmals dauert die Drucklagerung zu lange, um eine produktionsbegleitende Prüfung an 100% der zu testenden Objekte durchzuführen. Die Heliumkonzentration im Prüfobjekt hängt vom Druck bei der Lagerung, der Bombingzeit und dem freien Volumen im Inneren des Objekts ab. Zusätzlich spielt auch die Leckrate eine Rolle, die vor der Prüfung noch unbekannt ist. Nach Ende der Druckbeaufschlagung kann die Konzentration des Prüfgases im Inneren des Objektes nur berechnet, nicht aber gemessen werden. Sowohl nach Verschließen unter einer prüfgashaltigen Atmosphäre als auch nach der Drucklagerung wird das befüllte Prüfobjekt einige Zeit unter atmosphärischen Bedingungen gelagert. Dies ist erforderlich, um die Desorption von oberflächlich anhaftendem Helium zu ermöglichen. Wirkt auch nur ein kleiner Anteil der Materialien an der Oberfläche des Prüfobjekts als „Heliumschwamm“, wird die Empfindlichkeit der Methode vom Signaluntergrund beschränkt und die theoretischen Modelle werden versagen. Während der Wartezeit wird das Prüfgas aus dem Objekt durch die Leckkanäle diffundieren. Dies bedeutet, dass ein einmal gefülltes Objekt vor einer Prüfung nicht unbegrenzt gelagert werden kann. Demzufolge muss für jeden Bombingtest ein präzises Prüfrezept mit Zeitangaben entwickelt werden. In der Praxis werden oft Normen angewandt, in denen Klassen von Prüfobjekten definiert sind. Drücke und Lagerungszeiten werden in Abhängigkeit vom freien inneren Volumen des Prüfobjekts empfohlen. Die maximale Wartezeit darf für alle Produktklassen eine Stunde nicht überschreiten.
Tabelle 1: Parameter für die Drucklagerungsprüfung nach Mil-883E. Zusammenfassend wird während der Wartezeit die Nachweisgrenze durch Minimierung des Untergrundsignals verbessert. Die Fähigkeit zum Nachweis von Groblecks durch Gasverlust aus dem Prüfgasvorrat im Inneren des Prüfobjekts wird verschlechtert. Bei Blisterverpackungen kann das Prüfgas auch durch die angestochene Aluminiumfolie des Blisters mit einer Spritze eingebracht werden. Durch ein zweites Loch kann die eingeschlossene Luft entweichen und die Kavität gespült werden. Während der folgenden Prüfung werden die Löcher wieder abgeklebt. Diese Methode wird für absatzweise Prüfungen eingesetzt, um Fehler von Verpackungsmaschinen aufzudecken. Diese Methode ist zerstörend und kann nicht als produktionsbegleitende Prüfung eingesetzt werden. Im Falle von großen Leckagen kann das Prüfgas sehr schnell aus dem Prüfobjekt austreten. Schon während des Abpumpens der Vakuumkammer kann das gesamte Volumen der Kavität evakuiert werden und die hochempfindliche Helium-Dichtheitsprüfung wird blind gegen grobe Leckagen. Aus diesem Grund wird die Drucklagerungsprüfung meist als Feinlecktest eingesetzt, der mit einer Grobleckprüfung ergänzt wird. Diese zweite Methode kann eine Blasenprüfung, eine optische Prüfung oder jede andere Prüfmethode sein, deren Empfindlichkeitsbereich mit dem der Heliumprüfung überlappt. Eine Helium-Dichtheitsprüfung kann nicht ohne Weiteres eingesetzt werden, wenn die mechanische Stabilität des Prüfobjekts einen Differenzdruck von einem Bar (in der Kavität des Prüfobjekts) und Vakuum in der Prüfkammer nicht aushält. In diesem Fall muss das Prüfobjekt in der Kammer abgestützt werden. Typische Beispiele sind Lebensmittel- oder Blisterverpackungen.
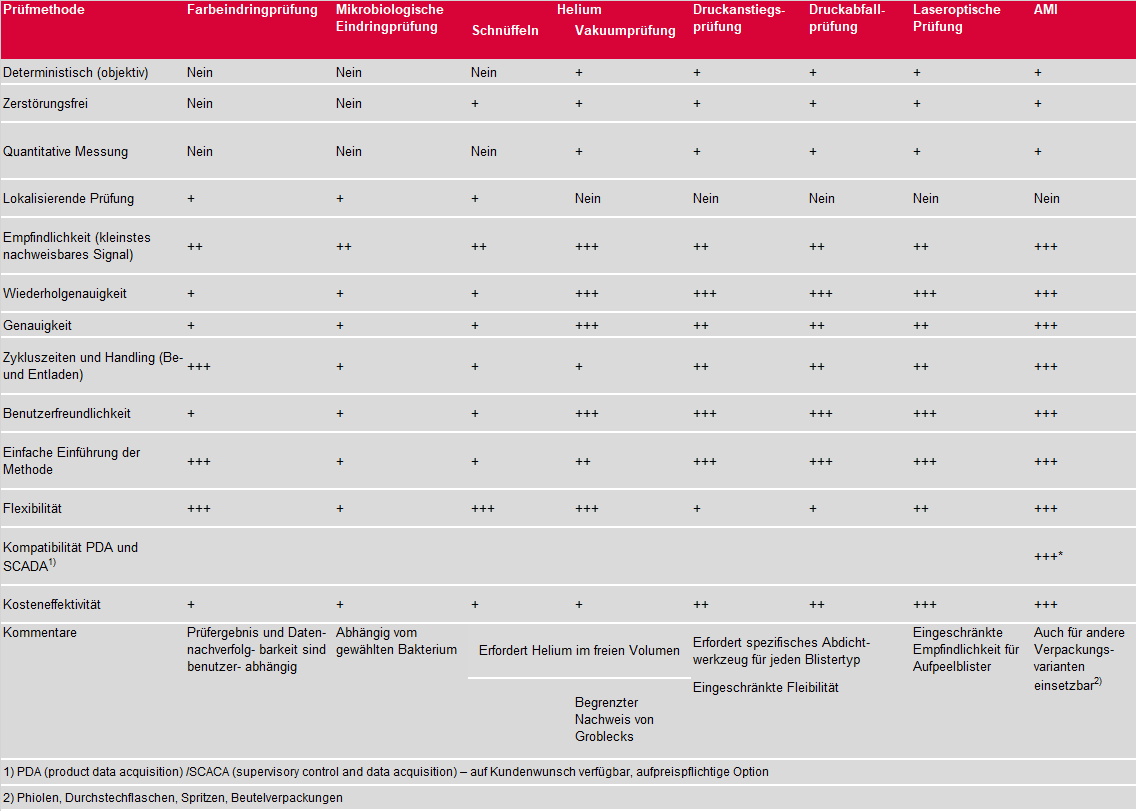
Der Weg zu einer neuen SensortechnologieDie Helium-Dichtheitsprüfung ist immer noch die empfindlichste Methode zur Prüfung hermetisch geschlossener Prüfobjekte. Wie bereits ausgeführt gibt es jedoch Beschränkungen, die meist mit der Zugabe des Prüfgases verbunden sind. Demzufolge war es notwendig, eine empfindliche Dichtheitsprüfung ohne spezifisches Prüfgas durchführen zu können. Als logische Konsequenz wurden Methoden entwickelt, die eine quantitative Dichtheitsprüfung mit dem bereits in der Kavität der Verpackung eingeschlossenen Gas durchführen. Als Prüfmethoden sind hier Druckänderungsprüfungen oder laseroptische Messungen zu nennen. Ein Überblick über die Prüfmethoden für die Unversehrtheit hermetisch geschlossener Prüfobjekte ist in Tabelle 2 gegeben: | ||
| Newsletter Mai 2023 VTU-Case-Study: Die Zukunft lebensrettender Medikamente gestalten - Mit einer hochmodernen Anlage, die in knapp zwei Jahren vom Konzept zur Marktreife entwickelt wurde. |
| |
Manuela Wagner, VTU Group GmbH, 25. Mai 2023Trotz Abklingen der Pandemie in Österreich entschied sich das Biotech-Unternehmen Biomay für die Errichtung einer neuen Anlage zur Herstellung von wichtigen protein- und DNA-basierten Inhaltsstoffen für die Medikamente von morgen. Nur 25 Monate nach der Entwicklung des ersten Konzepts konnte das Unternehmen seinen neuen, hochmodernen CDMO-Standort in der Nähe von Wien eröffnen. Dank seiner Entwicklungspartner und dem innovativen Konzept hinter dem 4.000 m² großen Hightech-Standort betreibt Biomay heute eine flexible Produktionsplattform für vielfältige Produkte in beliebigen Chargengrößen.
Der Ausdruck „lebensrettende Medikamente“ ist absolut keine Übertreibung. 2021 konnte das Unternehmen zum Beispiel seine Partnerschaft mit BioNTech SE zur Unterstützung der Lieferkette für die Herstellung des COVID-19-mRNA-Impfstoffs von Pfizer-BioNTech bestätigen.
Abbildung 1: Dr. Hans Huber, Chief Executive Officer von Biomay (Quelle aller Abbildungen: VTU Group GmbH).
Überwindung früher WachstumsschwierigkeitenAuch wenn sich Hans Huber heute zuversichtlich zeigt, war das nicht immer so. Vor einigen Jahren begegnete das Unternehmen einigen Wachstumsschwierigkeiten: Einerseits wurden die angefragten Chargengrößen und -typen immer unterschiedlicher und zerstückelter, einschließlich besonderer Anfragen nach kundenspezifischen Produkten. Andererseits brauchten auch einige Großkunden mehr Plasmid-DNA, das Ausgangsmaterial für mRNA, da die Produktion der COVID-Impfstoffe einfach zu dringend war. Als biopharmazeutische Contract Development and Manufacturing Organization (CDMO) arbeitet Biomay mit Kunden aus einem vielfältigen Mix internationaler Biotech-Unternehmen (Abb. 2). Bei einem maximalen Bioreaktorvolumen von nur 40 Litern waren der Produktionskapazität der älteren Anlage jedoch Grenzen gesetzt. „Das Anfragevolumen war einfach verrückt“, erinnert sich Hans Huber. „Der Zeitpunkt war gekommen, an dem wir eine Entscheidung über die zukünftige Ausrichtung unseres Unternehmens treffen mussten. Es war klar, dass wir umdenken, uns neu erfinden und erneut investieren mussten, um das nächste Level als CDMO zu erreichen“, so Hans Huber. „Unser Ziel ist es, führender Zulieferer von GMP-Leistungen zu sein, und damit die Bereitstellung von DNA-Plasmiden, Messenger-RNA und rekombinanten Proteinen zur Herstellung von bahnbrechenden Medikamenten zu sichern.“ 2018 entschieden Hans Huber und sein Team, Biomay zu erweitern und einen größeren Standort zu bauen. Als Begleiter bei diesem Prozess wählten sie eine internationale Technologie-Unternehmensgruppe mit breit gefächerter Erfahrung in der Ausführung umfangreicher CAPE-Projekte in den europäischen Life Sciences.
„Ich bin stolz, dass wir einen echten Beitrag leisten.“ DR. HANS HUBER, CEO, BIOMAY AG
„Ich erinnere mich gut an unsere erste Besprechung“, so Alexander Asbäck, Managing Director und Chief Operating Officer der VTU Group GmbH. „Wir trafen uns in einem Wiener Wohnviertel, wo die Techniker:innen in einem Labor zusammen saßen und unglaubliche Dinge mit Molekülen anstellten. Es war absolut klar, dass sie wirklich einen neuen, hochmodernen Standort brauchten – und auch verdienten.“ „Wir haben uns für die VTU nicht nur wegen ihrer Erfahrung bei EPCMv-Projekten entschieden, sondern auch, weil die Bereitschaft vorhanden war, an flexiblen Konzeptlösungen mit sehr knappem Budget zu arbeiten“, erläutert Hans Huber. „Das Team der VTU hat zugehört, mit uns an neuartigen Lösungen gearbeitet und war flexibel genug, unsere Bedürfnisse zu berücksichtigen und mit unseren Partnern zu kooperieren.“ In ihrer Rolle als EPCMv-Partner war die VTU verantwortlich für die Koordinierung des Projekts und die Bereitstellung der nötigen Expertise, die das Ausführungstempo erforderlich machte. Die nächste Frage war, wo, was und wie schnell errichtet werden sollte.
Abbildung 2: Biomay ist ein weltweit führender Komplettanbieter für GMP-Leistungen, der seinen anspruchsvollen internationalen Kunden DNA-Plasmide, Messenger-RNA und weitere Zellmaterialien liefert.
Gesamtheitlicher und nachhaltiger AnsatzBiomay gelang es, einen Standort und eine Baugenehmigung im Nordosten von Wien zu erwerben. Aspern Seestadt ist eines der größten städtischen Entwicklungsprojekte Europas, das für sein umweltfreundliches „grünes“ Profil bekannt ist. Abbildung 3: Upstream-Produktion im GMP-Reinraumbereich.
Bau eines zukunftssicheren CDMO-Standorts„Eine der ersten technischen Herausforderungen war die Festlegung der Spezifikationen. Diese mussten eine für die Ausführung von vielschichtigen Prozessen nötige Flexibilität bieten, ein zukunftssicheres Konzept unterstützen und GMP-konform sein (Abb. 4) – und all das, ohne ein knapp berechnetes Budget zu sprengen“, so Alexander Asbäck, „alles drehte sich um Flexibilität.“
Abbildung 4: Qualität in allen Details. Das vielseitige Gebäude ist täuschend einfach gehalten und bietet alles, was für einen Fertigungsstandort der nächsten Generation nötig ist. Die GMP-Fertigungskonfiguration folgt einem klassischen „Upstream-Downstream“-Ablauf, jedoch mit einem Unterschied: Hier gibt es auch einen speziellen Bereich für neuartige und individuelle (patient:innenspezifische) Chargen.
„Alles drehte sich um Flexibilität.” ALEXANDER ASBÄCK, CHIEF OPERATING OFFICER, VTU ENGINEERING
Im Upstream mit einem Edelstahl-Gärungsbehälter von 750 Litern verzehnfachte Biomay seine Kapazitäten im Hinblick auf Chargengrößen und -volumen. Das Unternehmen verfügt jetzt über Bioreaktoren mit einer Bruttokapazität von 5, 50, 150 und 750 Litern (Abb. 5). Das Vorlauf-Layout ist in mehrere Reinraumbereiche unterteilt und umfasst eine zentrale Medienversorgung sowie Separierung, Zellspaltung durch Homogenisierung oder chemische Lyse und Sedimentation. Die Reinigungsvorgänge im Downstream bestehen aus einer Chromatografie, auf die durch Panel und mobile Behälter flexibel konfigurierbare Ultrafiltrationsgestelle folgen. Der Bereich für die sterile Abfüllung ist in einem eigenen Reinraum untergebracht. VTU war an allen Vorgängen beteiligt: von Machbarkeitsanalyse und Konzept über Beschaffung und Bau bis zu Dokumentation und Abnahme.
Patient:innenspezifische BehandlungenEin spezieller Aspekt des Konzepts ist Biomays Anlage zur Herstellung individueller Fertigungschargen für patient:innenspezifische Medikamente. „Stellen Sie sich das einmal vor: Wir können in nur wenigen Wochen individuelle Medikamente für Krebspatient:innen in GMP-Qualität herstellen“, freut sich Hans Huber. Gemeinsam mit seinen Kunden liefert Biomay nun DNA-Plasmide für patient:innenspezifische – auch als „Neoantigene“ bezeichnete – Tumor-Antigene als neue, vielversprechende Lösung zur Heilung von Krebspatient:innen. „Dies erfordert von uns viel Innovationskraft und Kreativität, um die knappen Zeitrahmen einzuhalten und trotzdem kosteneffizient zu bleiben“, so Hans Huber.
Abbildung 5: Moderner Bioreaktor mit digitaler Echtzeitüberwachung.
Bauen während einer PandemieDie Planung und der Bau des Standorts dauerten 25 Monate von Ende 2019 bis Ende Januar 2022. Eine der großen Herausforderungen stellte die Covid-Pandemie dar, daher fanden die meisten Planungsbesprechungen von Biomay, VTU und allen anderen Beteiligten online statt. „Zusätzlich zu allen anderen Anforderungen hatten die Sicherstellung eines sicheren und gesunden Arbeitsbereichs und die Einhaltung einer ‚Zero Accident‘-Richtlinie für uns oberste Priorität“, betont Thomas Miklautsch, Managing Director von VTU Engineering, der das Projekt sehr eng betreute. Da es sich um ein für das öffentliche Gesundheitswesen wichtiges Projekt handelte, wurden sehr früh Impfungen vorgenommen und Präsenzbesprechungen auf ein Minimum reduziert. „Ironischerweise haben wir mitten in der Pandemie einen Standort zur Herstellung des Covid-Impfstoffs gebaut“, so Thomas Miklautsch. „Letztendlich hat Biomay von uns ein Komplett-Package bekommen – vom ersten Entwurf bis zur abschließenden Zertifizierung“, freut sich Miklautsch. „Dabei haben wir den Liefertermin und die Budgetvorgaben eingehalten und die modernsten Planungstools verwendet.“ Eine breite Kompetenzenpalette, fortschrittliche Entwicklungsplanungstools und eine unkonventionelle Denkweise gehörten zu den wichtigsten Erfolgsfaktoren.
„Unsere digitale Entwicklungsplattform war für die Projektorganisation unerlässlich.” GLORIA GALINDO-PEITBUCHNER, SENIOR PROCESS ENGINEER, VTU ENGINEERING Digitale EntwicklungsplattformEine weitere Prozess-Expertin, die einbezogen wurde, war Gloria Galindo-Peitbuchner, Senior Process Engineer bei VTU. Für sie war die virtuelle Arbeit Neuland und etwas gewöhnungsbedürftig. Eine große Herausforderung war auch das „Smart Working“ zur Festlegung effizienter Engineering-Standards, die es bei Biomay aufgrund des schnellen Wachstums und der Unternehmensgröße noch nicht gab. „Unsere digitale Entwicklungsplattform hat zur Projektorganisation beigetragen“, so Gloria Galindo-Peitbuchner. „Sie stellte die Struktur bereit, um schnell voranzukommen und alle GMP- und weiteren wichtigen Zertifizierungen zu erhalten.“
Standardisierte ProduktionsprozesseUm eine „Überdimensionierung“ zu vermeiden und nicht bei jeder geringfügigen Konzeptänderung der Prozesse eine Freigabe einholen zu müssen, entschied sich Biomay für standardisierte Biotech-Produktionsprozesse. Das beinhaltete auch die Wahl von Standard-Edelstahlvorrichtungen – statt Einweggerätschaften mit Einwegartikeln –, mit denen die Mitarbeiter:innen im Hinblick auf die CIP- und SIP-Verfahren zur Sterilisation vertraut waren. Der Standort erhielt kein „Ballroom-Konzept“, sondern wurde in kleinere Räume unterteilt, um die Produktionswechselzeiten zu minimieren (Abb. 6). Für die Zukunft erwartet Hans Huber einen größeren Produktionsmaßstab, auch für klinische Studien der 3. Phase oder für die Markteinführung neuer Produkte. „Wir verfügen jetzt über einen Standort, der uns die Möglichkeit eines strategischen Wachstums und unseren Kunden attraktive Produktionskapazitäten bietet”.
Abbildung 6: Bioreaktoren und weitere Vorrichtungen sind mit Rollen versehen, um flexible Chargenkonfigurationen und eine einfache Reinigung zu ermöglichen.
Kurzinfos zu Biomay
Kapazitäten und Produkte
Wichtigste EPCMv-Erfolgsfaktoren
Fünf maßgebliche Entwicklungsherausforderungen
Wichtigste EntwicklungspartnerArchitekt:innen: Delta Podsedensek Architekten ZT GmbH HVAC / Bauleistungen: TECH.CON GmbH Anlagen- und Process Engineering: VTU Engineering GmbH Prozessautomatisierung: WK-Tech GmbH Kontakt:VTU Group GmbH
| ||
| Newsletter April 2023 SPETEC-Reinraumkonzept bietet Apurano beachtlichen Innovationsvorsprung |
| |
Elisabeth Schollwöck, SPETEC GmbHDer Life Sciences Newcomer Apurano hat ein einzigartiges Verfahren zur Herstellung und Abfüllung von Arznei- und Nahrungsergänzungsmitteln entwickelt. Nun will das Unternehmen mit seinen innovativen Gesundheitsprodukten einen Teil dieses dynamisch wachsenden Marktes erobern. Die hierzu erforderliche Produktionsumgebung konnte optimal und in kurzer Zeit mit einem modularen Reinraumkonzept von SPETEC realisiert werden. Die Zahl der Allergiker steigt, immer mehr Menschen leiden unter Autoimmunerkrankungen, Fälle von Neurodermitis, Asthma und Psoriasis ebenso wie Colitis Ulcerosa oder Morbus Crohn nehmen weiter zu. Daneben nehmen in Deutschland täglich 13,5 Mio Menschen Vitamine als Nahrungsergänzungsmittel zu sich. Mit einem neuartigen und patentierten PuranoTec®-Verfahren löst Apurano nunmehr das alte Problem der Naturheilkunde: Viele Inhaltsstoffe von wirksamen Heilpflanzen sind nicht wasserlöslich und können daher nur zu einem sehr geringen Teil über den Dünndarm aufgenommen werden. Hierbei spielt auch die Größe der Partikel eine wesentliche Rolle, da der Dünndarm die Eigenschaft eines Filters hat und nur durchlässig für Partikel kleiner als 5 μm ist. Somit sind viele der am Markt verfügbaren Heilpflanzenprodukte in magensaftresistenten Kapseln nur in geringem Ausmaß wirksam, da hieraus maximal 5 % der wasserunlöslichen oder schwer wasserlöslichen Bestandteile aufgenommen werden können. Uraltes Problem mit High-Tech gelöstApurano entwickelt und produziert High-Tech-Phytoceuticals, die schwer wasserlösliche Bestandteile von Heilpflanzen oder Vitalpilzen mittels eines physikalischen Verfahrens in eine für den menschlichen Körper optimal resorbierbare flüssige Form bringen und dabei vollständig ohne Chemie oder Konservierungsstoffe auskommen. So werden Produkte mittels eines Aktiv-Sprays in die Wangeninnenseite und unter die Zunge gesprüht, wo alle Bestandteile der Pflanze oder des Pilzes aufgrund ihrer speziellen Oberflächenbeschaffenheit optimal über die Mundschleimhaut resorbiert werden. Eine Verstoffwechselung über die Leber wird überflüssig und die Aufnahme der naturheilkundlichen Produkte lässt sich verbessern. Damit erhöht sich einerseits die Wirksamkeit der Produkte, andererseits sinken die Kosten für den Anwender. Zur Herstellung solch innovativer Medizinprodukte, Nahrungsergänzungsmittel und Arzneimittel nach GMP benötigte das junge Unternehmen für sein neues Firmengebäude eine leistungsfähige Reinraumumgebung zur gesicherten, anspruchsvollen Verarbeitung und Abfüllung der Medizinalpilze und Heilpflanzen. Dr. Werner Brand, Geschäftsführer von Apurano und zugleich Entwickler des patentierten Verfahrens, entschied sich für einen Reinraum-Anbieter, der u. a. auch flexibel auf die in der Bau- und Installationsphase auftauchenden Herausforderungen eingehen konnte. Nach intensiver Recherche fiel die Wahl schließlich auf den in Erding bei München ansässigen Reinraumspezialisten SPETEC. Modularität der ReinraumkomponentenDabei erwies sich insbesondere die Modularität der von Spetec angebotenen Reinraum-Komponenten als großer Vorteil gegenüber anderen Anbietern. Auch die räumliche Nähe und Flexibilität der SPETEC-Experten innerhalb des gesteckten Zeitplans fielen ins Gewicht. Das bei Apurano installierte Laminar-Flow-Box-System wurde als Raum-in-Raum-Lösung realisiert, wobei der gesamte Reinraumbereich rund 25 m2 groß ist und verschiedene Zonen umfasst. Dies beginnt beim Entkeimungsraum (Klasse D), in den das Rohmaterial durch eine erste Schleuse aus dem Wareneingang eingebracht wird, um dort mit einem sehr speziellen und streng geheimen Verfahren zerkleinert und entkeimt zu werden.
Durch eine zweite Schleuse wird das aufbereitete Rohmaterial dann aus einem Tank im Entkeimungsraum über ein aufwändiges Schlauchsystem in einen weiteren Raum (Klasse B) gepumpt. In diesem zweiten Raum wiederum ist eine Reinraumzelle der Klasse A installiert, in der sich die Abfüllanlage für das fertige Produkt befindet. Innerhalb dieses Klasse-A-Umfelds wird die kleine Anwendungsflasche mit der Wirklösung befüllt und der Spezial-Sprühkopf montiert. Aus dieser Zelle wird die Flasche dann über eine weitere Schleuse hinaus in die Etikettierung transportiert. Um diese Beförderung der abgefüllten Flaschen berührungslos zu gestalten, wurden an der Übergabestation 2 gegenläufig rotierende Drehteller installiert, welche die kleinen Flaschen allein durch die entstehenden Zentrifugalkräfte vom einen zum anderen hinüber befördern. In Kombination mit einem SPETEC® Laminar Flow Modul FMS, das u. a. über einen Hochleistungsfilter des Typs H14 verfügt, lässt sich ein Isolationsfaktor von 10 000 erreichen. Damit verbessert sich die Luftqualität gegenüber der Umgebung mindestens um das 10 000-fache.
Abbildung 1: Die installierte Füll-und-Verschließstation (Quelle aller Abbildungen: Spetec GmbH).
Daneben wird die Partikelkonzentration innerhalb der Einheit von rund 15 Mio. auf etwa 1 500 Partikel pro m3 reduziert. Im Ergebnis wird eine nach DIN-ISO-zertifizierte Reinraumklasse 5 erreicht (nach US Fed. 209E die Klasse 100). Mit dieser ausgefeilten Raum-in-Raum-Lösung konnte der Reinraumbereich innerhalb der baulichen Hülle noch GMP-konformer gestaltet werden. Für die Genehmigung dieses modularen RR-Konzepts durch die Regierung von Oberbayern war dies unerlässlich.
Abbildung 2: Die Raum-in-Raum-Lösung.
Zuverlässige Produktion schon während der Qualifizierungsphase Dass heute alle Komponenten zuverlässig und ohne Schwierigkeiten funktionieren und die Produktion die geplante Auslastung von 2 400 Flaschen pro h bei einer Verfügbarkeit von 98 % – also quasi unterbrechungsfrei – erreicht, weiß Apurano-Geschäftsführer Dr. Brand ebenso zu schätzen, wie die reibungslose Zertifizierung des Reinraumsystems durch SPETEC. Vor allem, „dass schon während der Qualifizierungsphase die Spezifikation zu 100 %erfüllt wurde“ sieht Brand als großen Vorteil. Davor, dass die Expansionsfalle zuschnappen und die Kapazitätsgrenzen aufgrund hoher Marktnachfrage rasch erreicht werden könnten, hat Brand keine Angst. „Wir können sehr rasch expandieren, das Grundstück und die Pläne haben wir bereits. Und den passenden Reinraum-Lieferanten auch“, sagt Brand mit Blick auf SPETEC und das zurückliegende, gelungene Reinraumprojekt. Kontakt: | ||
| Newsletter Februar 2023 Keine erfolgreiche CCS ohne systematisches QRM? |
| |
Simon Fiala, comprei Reinraum-Handel- und Schulungs GesmbH
AusgangslageEin Blick auf die gelebte Praxis im pharmazeutischen Umfeld zeigt: Etablierte Maßnahmen der Kontaminationskontrolle und deren Dokumentation liegen oftmals in sehr heterogener Tiefe und teilweise unzureichender Konsistenz vor. Eine nähere Betrachtung macht sichtbar, dass die betreffenden Dokumente und die dargelegten Maßnahmen vielfach kaum mit dem tatsächlichen Fehlerpotential bei der Herstellung steriler Arzneimittel korrespondieren. Immer wieder ergibt sich das Bild eines Cocktail-Mix aus empirischen Daten und regulativen Anforderungen, gefärbt von subjektiven Wahrnehmungen und Eindrücken – anstatt einer systematischen und nachvollziehbaren Vorgehensweise aufgrund einer risikobasierten Betrachtung und entsprechenden Ableitung. Und genau dieses Spannungsfeld scheint die neue Version des Annex 1 (Manufacture of Sterile Medicinal Products) des EU-GMP-Leitfaden aufzugreifen. Die Leitlinie betont, wie Hersteller neue Möglichkeiten nutzen können, die sich aus der Anwendung eines verbesserten Prozessverständnisses ergeben, indem innovative Werkzeuge verwendet werden, wie unter anderem in den ICH-Guidelines Q9 „Quality Risk Management“ und Q10 „Pharmaceutical Quality System“ beschreiben. Als zentrales Ziel das Annex 1 in der neuesten Version kann die Sicherstellung adäquater Kontaminationskontrolle verstanden werden (Sterility Assurance, Bioburden, Pyrogene, Partikel) ? basierend auf einem genauen Verständnis der Art des Produkts und der Prozesse. Für den Anwender wächst somit die Notwendigkeit, geeignete Tools des Risikomanagement aus einem umfassenden Methodenkoffer auszuwählen und gezielt einzusetzen – im Besonderen für die Etablierung einer nun dezidiert geforderten Kontaminations-Kontrollstrategie (Contamination Control Strategy, CCS). Selbstverständlich unterstützen diese Werkzeuge auch vielfältige weitere Belange im Spannungsfeld der pharmazeutischen Qualitätssicherung und des gelebten Qualitätsmanagements. Der Einsatz etablierter Tools für die systematische Determinierung bestehender Risken soll ein solides Fundament für das Setup einer umfassenden übergeordneten Kontaminations-Kontrollstrategie bilden. Anders ausgedrückt kann die These erhoben werden: Es ist keine erfolgreiche Kontaminations-Kontrollstrategie ohne systematisches Qualitätsrisikomanagement möglich. | ||
| Newsletter Januar 2023 Pharmaceutical Robots: Third-party testing to meet GMP requirements for pharmaceutical Manufacturing |
| |
Dominique Schmeer, 31. Januar 2023Aseptic robots are increasingly replacing manual production in the pharmaceutical industry. Stäubli partnered with SKAN in the development of its Stericlean robots in order to optimise product design. The article shows the advantages of independent third-party testing for both the manufacturer and the customer. The pharmaceutical industry has slowly but steadily shifted from a human-centric environment to a fully automated one. Ever stricter requirements regarding hygiene, safety, and productivity are all pushing the industry away from manual production, and the COVID-19 pandemic has accelerated this change. Recently, regulatory requirements have become even more restrictive with the revision of EU GMP Annex 1: “Manufacture of Sterile Medicinal Products” (25 Aug 2022). The use of robotics is mentioned in Section 2 with the following wording: "Robotic systems … should be considered to increase the protection of the product from potential extraneous sources of … particulate and microbial contamination such as personnel …" According to the new Annex 1, robotic systems are finding their place in this sensitive area of sterile pharmaceutical manufacturing. Stäubli Stericlean, which launched in 2009, was the first range of robots to fulfil aseptic operation requirements as mandatory for working in an ISO 5/Grade A environment, particularly for use inside isolators and restricted access barrier systems (RABS).
An opportunity for a more complete GMP compliance documentation packageTo meet the new GMP requirements and further develop the quality of its Robotic portfolio, Stäubli entered into a partnership with SKAN, the leading manufacturer of isolator and aseptic processing systems and specialists in pharmaceutical and aseptic environments, in Aug 2021. SKAN’s analytical services (SKANalytix) helped Stäubli to improve the design of its robots for use in an aseptic environment and enhance the cleanability, hygienic design, and surface decontamination capabilities with vaporised or nebulised hydrogen peroxide, alongside other important considerations (detailed below) that make the robot suitable for installation in the highest pharmaceutical cleanroom class ISO 5/Grade A for aseptic manufacturing. All the tests aim to provide the robots with the much-needed GMP compliance documentation package that customers demand.
Independent, third-party analytical supportThe SKANalytix service provides independent, third-party analytical support for questions and concerns related to aseptic processing and isolators and also produces studies and conducts tests to evaluate the pharmaceutical process safety and operator safety of the robots in isolator and cleanroom technology. The collaboration between Stäubli and SKAN has led to the development of new features such as the hygiene design in critical areas or the design of the sealing for the Stericlean package, which ensures that the robots are suitable for aseptic manufacturing conditions. Hygienic design is key aspect to ensure that a robot is suitable for working in an aseptic environment. The requirements for the design are manifold.
GMP requirements for robots in an aseptic environmentTwo main factors are particularly important: Firstly, it must be possible to properly clean and decontaminate the entire outer surface of the robot. Otherwise, there is a risk that the aseptic processing conditions cannot be maintained. The surface material and gasket materials must be chosen accordingly, and the design must allow for easy access and cleaning. The second main factor is the moving parts, which pose the greatest risk for particle generation. Therefore, special attention needs to go into the design and the sealing of joints. In particular, there should be no gaps in the joints of the robot’s outer shell where microorganisms could accumulate and grow. The surface roughness Ra must not exceed 0.8 µm to prevent fungi or bacteria from colonising and to allow for efficient cleaning. The surface must withstand all cleaning and surface decontamination procedures, especially vaporised hydrogen peroxide (H2O2), which is used for surface decontamination inside isolators. A robot should not generate turbulence in the unidirectional airflow when in motion. Particles released during movement must not exceed a defined low threshold to ensure compliance with ISO 5/Grade A standards. Lastly, all areas must be accessible so that the robot can be easily cleaned and decontaminated and no substances, particles, or microorganisms can accumulate, build up, or pool. (Fig. 2 and 3 show the cleaning performance. Fig. 4 shows the test performance according to ISO 5/Grade A requirements.)
Intensive testing for robot cleanability, resistance, and movement in an isolatorThe Stäubli Stericlean TS2-60 and TX2-40, -60, and -90 robots have been subjected to SKANalytix’s intensive testing to determine what improvements could be made to further tailor the design to the cleanroom isolator environment. As an example, robocell – the first fully EU-GMP-Annex-1-compliant, fully robotic aseptic filling line, which was a collaboration between groninger and SKAN – also benefited from the tests (fig. 1). The robocell received the 2022 ISPE DACH Future Robotic Award for their GMP Annex 1 Compliance Robotic integration.
Figure 1: Robocell fully robotized aseptic filling (Source: SKAN) Each test provides information about a specific aspect of the robot design. Altogether, they provide a complete picture of a robot and its suitability for use in an isolator. The individual tests take between a few hours and several weeks. The procedure for each test is briefly described below:
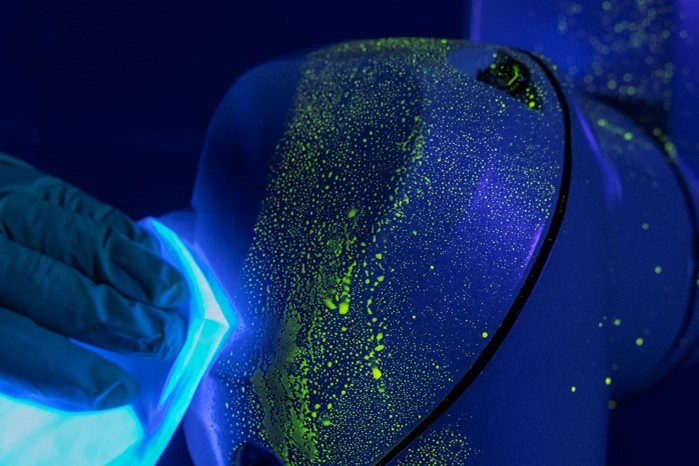
1. Cleanability of the equipmentThe robot is sprayed with a fluorescent tracer substance, e.g. riboflavin, before being cleaned. After cleaning, the remaining fluorescence helps identify weaknesses in the design. It shows where accessibility is insufficient, and contamination is difficult to remove or could build up. Testing can go further by precisely spiking surrogate contaminations and detecting them down to the nanogram level after the cleaning procedure (fig. 2 and fig. 3).
Figure 2: A Stäubli robot with fluorescent tracer substance (riboflavin)(Source: Stäubli/SKAN).
2. Chemical and microbiological resistanceThese tests focus on ensuring that all materials resist a panel of commonly used cleaning and decontamination agents, including vaporized or nebulized Hydrogen Peroxide (H2O2) All materials must also be inert to bacteria and mould. A standardised set of microorganisms is inoculated onto the construction material, and their growth is monitored over four weeks. 3. Adsorption/DesorptionMaterials should not adsorb H2O2 during decontamination procedures. When adsorption is strong or outgassing is slow, it can result in H2O2 concentrations inside an isolator that can be harmful to the product being handled. The decontamination process can also become unnecessarily long. SKAN practices standardised testing of H2O2 adsorption and desorption kinetics. 4. D-value testsIn microbiology, D-value is the amount of time required to achieve a 1(log) reduction (or 90% reduction) in bacteria under a given set of conditions. A low D-value equates to faster and easier decontamination. The efficiency of H2O2 decontamination can vary according to the material. Different finishings on a robot may influence how contamination is deposited on a surface, or how easily H2O2 neutralises that contamination.
| ||




 Zusätzlich zu hochmodernen Technologien ist ein nachhaltiges Konzept ein wichtiger Aspekt dieses Standorts, der über eine Grundwasser-Wärmepumpe zur Heizung und Klimatisierung des gesamten Gebäudes sowie Solarpanele zur Erzeugung von Strom verfügt (Abb. 3).
Zusätzlich zu hochmodernen Technologien ist ein nachhaltiges Konzept ein wichtiger Aspekt dieses Standorts, der über eine Grundwasser-Wärmepumpe zur Heizung und Klimatisierung des gesamten Gebäudes sowie Solarpanele zur Erzeugung von Strom verfügt (Abb. 3).





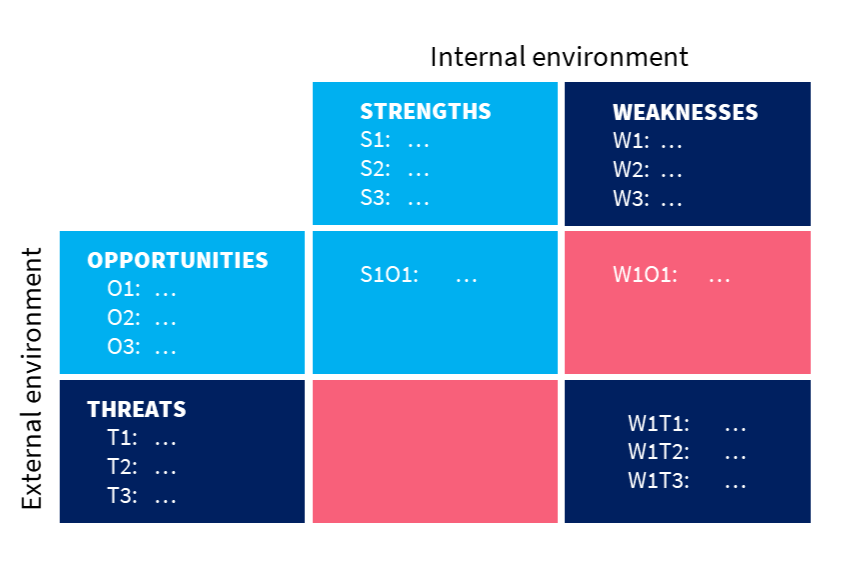 Opportunities und Threats – Stärken, Schwächen, Chancen und Risiken) stellt im Kontext des gegenständlichen Vorhabens ein Instrument der strategischen Umsetzbarkeits- und Planungsbewertung dar. Sie dient somit der Positionsbestimmung und der Strategieentwicklung für die Umsetzung der systematisch definierten Maßnahmen. Die SWOT-Analyse kann auch in systematischer Verknüpfung mit einer TOWS-Matrix herangezogen werden. Hinter TOWS steckt nicht nur die Neuordnung des Akronyms SWOT. Hier kann der Nachteil der rein qualitativ orientierten SWOT-Analyse durch das Verknüpfen mit der quantitativ orientierten TOWS-Matrix neutralisiert werden. Dieses Mixed-Methods-Model bietet die Möglichkeit, einen kennzahlenbasierenden Output zu gewinnen, ohne an Kreativität und gestalterischer Freiheit im Zuge der Analyse zu verlieren.
Opportunities und Threats – Stärken, Schwächen, Chancen und Risiken) stellt im Kontext des gegenständlichen Vorhabens ein Instrument der strategischen Umsetzbarkeits- und Planungsbewertung dar. Sie dient somit der Positionsbestimmung und der Strategieentwicklung für die Umsetzung der systematisch definierten Maßnahmen. Die SWOT-Analyse kann auch in systematischer Verknüpfung mit einer TOWS-Matrix herangezogen werden. Hinter TOWS steckt nicht nur die Neuordnung des Akronyms SWOT. Hier kann der Nachteil der rein qualitativ orientierten SWOT-Analyse durch das Verknüpfen mit der quantitativ orientierten TOWS-Matrix neutralisiert werden. Dieses Mixed-Methods-Model bietet die Möglichkeit, einen kennzahlenbasierenden Output zu gewinnen, ohne an Kreativität und gestalterischer Freiheit im Zuge der Analyse zu verlieren.

 5. Particle emissions
5. Particle emissions
