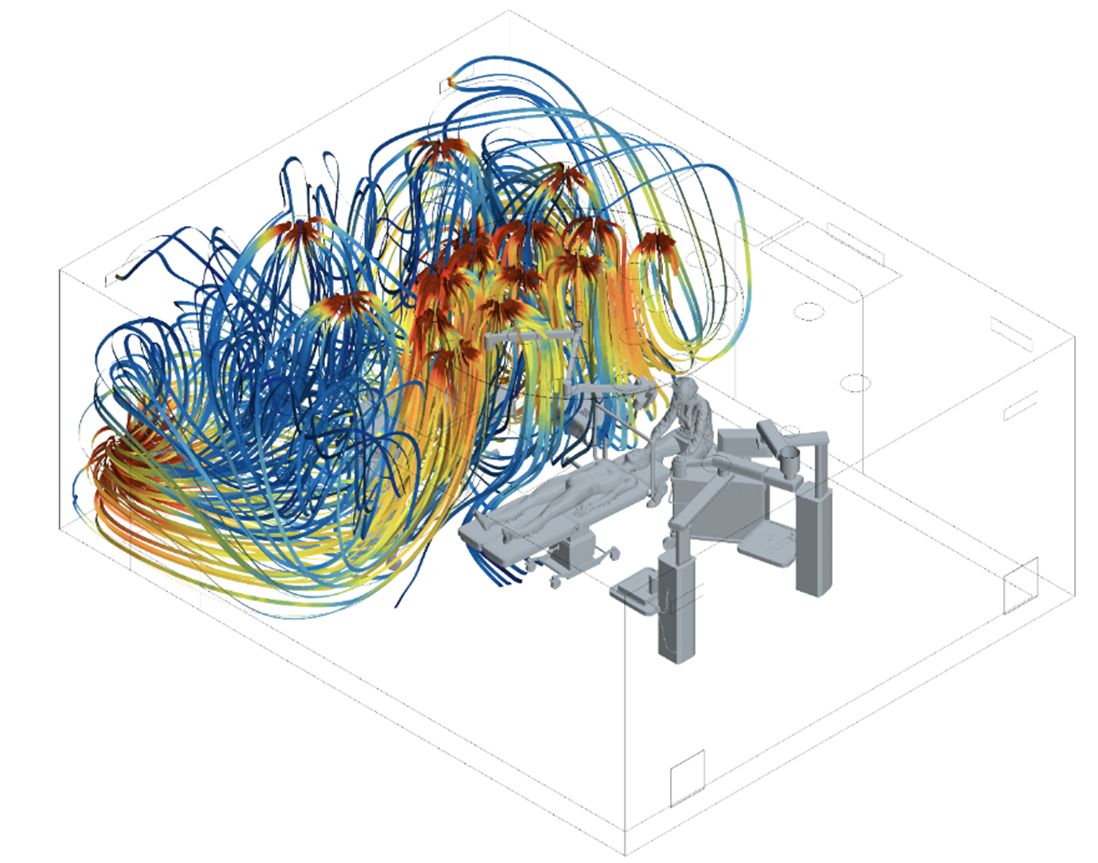
Hohe Sauberkeitslevel stabil erreichen | Beitrag aus cleanroom & processes 5 | Nr. 2 | 86-91 (2026)
31.07.2026Für eine zukunfts- und konkurrenzfähige Position im Markt strukturieren immer mehr Unternehmen ihr Produktangebot neu. Der Trend geht zu anspruchsvollen Lösungen für Hightech-Branchen. Verbunden ...
Ausbau des SÜDPACK-Standorts in Coulmer abgeschlossen
31.07.2026Der 17. Juli 2026 markiert einen wichtigen Meilenstein in der Geschichte der SÜDPACK Medica: In Anwesenheit zahlreicher örtlicher Honoratioren, Projektbeteiligter, Nachbarn, Mitarbeitenden, der ...
Steriline präsentiert OmniAI Vision und OptiLAIN
29.07.2026Steriline präsentiert OmniAI Vision und OptiLAIN: eine neue Generation intelligenter Technologien, die den Prozess gemäß den Anforderungen von Anhang 1 und Anhang 22 beobachten, verstehen und ...
Vitrealab baut Reinraum aus, um laser-LCoS-Lichtmaschinen für AR hochzufahren
28.07.2026Der deutsche Photonik-Startup Vitrealab erweitert seine Reinraumkapazitäten, um die Produktion von laser-illuminated LCoS-Lichtmaschinen für Augmented-Reality-Brillen deutlich zu steigern. Die ...
CRC stärkt Führungsteam und expandiert international: Wachstumskurs als Antwort auf steigende Nachfrage in Halbleiter-, Photonik- und Life Sciences-Branche
27.07.2026Die CRC Clean Room Consulting GmbH setzt ihren Wachstumskurs konsequent fort und positioniert sich strategisch für internationale Märkte. Mit der Verstärkung ihres Managementteams, dem Ausbau der ...
Branchenpartner
meistgelesen

Beitrag aus der Ausgabe 2/2026 der Zeitschrift cleanroom & processes
Hohe Sauberkeitslevel stabil erreichen
Mehr als nur ein Reinigungsprozess
Die industrielle Wertschöpfung befindet sich im Umbruch. Bisherige Absatzmärkte verändern sich, neue Technologien erfordern angepasste Geschäftsmodelle und Produkte. Um ihre Wettbewerbsfähigkeit zu erhalten bzw. zu stärken, setzen immer mehr Unternehmen auf die Fertigung hochwertiger Produkte und Komponenten mit guten Margen. Der Fokus liegt auf Hightech-Branchen, z. B. Halbleiter-Zulieferindustrie, Elektronikfertigung, E-Mobility, ...

Beitrag aus der Ausgabe 2/2026 der Zeitschrift cleanroom & processes
Die saubere Zelle
Weniger Luftwechsel aber mehr Reinheit?
In der Medizin sind sie längst angekommen – zellbasierte Therapeutika. Doch ihre Produktion stellt die Hersteller noch immer vor Herausforderungen, da die Zellprodukte nicht terminal sterilisiert werden können. Ein Team um den japanischen Ingenieur Masahiro Kino-oka zeigt nun, wie sich die Reinheit der aseptischen Kernzone durch cleveres Lüftungsdesign optimieren lässt.Es klingt immer noch ein bisschen nach Science-Fiction: Einem ...

Beitrag aus der Ausgabe 2/2026 der Zeitschrift cleanroom & processes
Schnee statt hochreinem Wasser
Medizin- und Pharmaprodukte sicher und ressourcenschonend reinigen
Das Spektrum an medizintechnischen Produkten und pharmazeutischen Erzeugnissen, die verpackt werden müssen, ist enorm vielfältig und stellt besondere Anforderungen an die Reinigung während der Herstellung. Dabei ist ein produktspezifisch definiertes Reinheitslevel zuverlässig zu erfüllen. Hinzu kommen regulatorische Vorgaben, die eingehalten werden müssen. Entsprechend ist ein Reinigungsprozess dann für die Herstellung von Medizintechnik- und ...
Top Themen
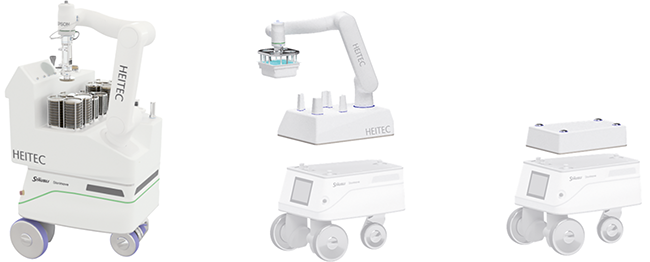
Beitrag aus der Ausgabe 2/2026 der Zeitschrift cleanroom & processes
Innovationen im pharmazeutischen Reinraum
Kontaktlose Authentifizierung, mobile Robotik und intelligente Bedienkonzepte für die Pharmaproduktion
In hochreinen Produktionsbereichen der Pharmaindustrie steht die Vermeidung von Kontaminationen an oberster Stelle. Das größte Risiko geht dabei vom Menschen aus, weshalb Reinräume nur von autorisiertem Fachpersonal betreten werden dürfen. Gleichzeitig erfordern moderne Herstellprozesse eine lückenlose Kontrolle und Dokumentation, um Qualität und Compliance – etwa mit den GMP-Regularien – sicherzustellen. Traditionelle Verfahren ...

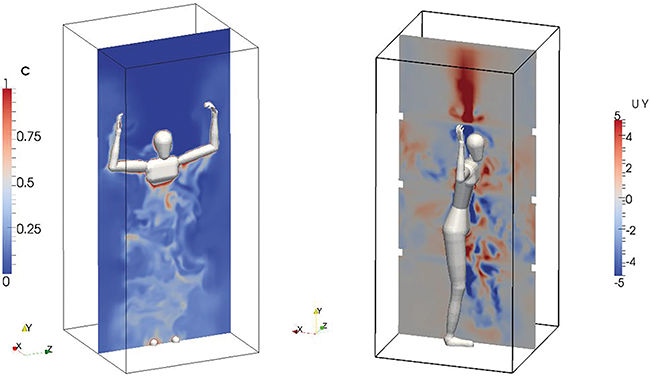
Vorschau (Änderungen vorbehalten)

Beitrag aus der nächsten Ausgabe 03/2026 der Zeitschrift cleanroom & processes
(erscheint am 18.09.2026)
Trockenraum und Mensch - Wechselwirkungen und Beeinflussung
Die Produktion von Batteriezellen erfordert Rein- und Trockenräume mit hoher Partikelreinheit und extrem niedriger Luftfeuchte. Während die Technik solcher Umgebungen gut beschrieben ist, ist der Einfluss des Personals auf den Taupunkt sowie die Rückwirkung des trockenen Klimas auf die Mitarbeitenden kaum untersucht. Im Rein- und Trockenraumlabor des Fraunhofer IPA wurden Versuche zur durch Personen verursachten Veränderung des Taupunkts durchgeführt. Auf Basis der Ergebnisse wurden Verhaltensregeln sowie organisatorische und technische Maßnahmen abgeleitet, die sowohl die Stabilität des Trockenraumklimas als auch den Gesundheitsschutz unterstützen.